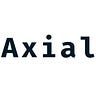


Axial Discovery - High fidelity CRISPR
Call for talented individuals and teams

Analysis sent out every Monday
Build with Axial: https://axial22.axialvc.com/
Axial partners with great founders and inventors. We invest in early-stage life sciences companies such as Appia Bio, Seranova Bio, Delix Therapeutics, Simcha Therapeutics, among others often when they are no more than an idea. We are fanatical about helping the rare inventor who is compelled to build their own enduring business. If you or someone you know has a great idea or company in life sciences, Axial would be excited to get to know you and possibly invest in your vision and company . We are excited to be in business with you - email us at info@axialvc.com

High fidelity CRISPR
CRISPR gene editing is one of the hottest fields in biotech right now. With the promise to cure genetic diseases, the field has attracted some of the most talented scientists in the world. But this promise has also attracted a large group of stock promoters that have essentially made some CRISPR companies & their securities quasi-religions temporarily. Nevertheless, gene editing is set up to transform medicine by providing potential cures. The field got started in 2012 from work led by Emmanuelle Charpentier and Jennifer Doudna who established CRISPR as a tool to edit genes and earned the Nobel Prize for it. This publication was part of an annus mirabilis in life sciences that also saw the advent of single-cell sequencing, CAR-T’s success, and more. The quote-on-quote B.C. era (before 2012) of gene editing was built upon TALENS (it took me a year to clone one in college) and zinc-finger nucleases (ZFN). After 2012, a gold rush for CRISPR proteins began. The first versions were similar to scissors and sparked the formation of Editas, Caribou, Intellia, ERS, Inscripta, and CRISPR Therapeutics. Version 2, centered around base editing, is more similar to an eraser with Beam Therapeutics along with Verve commercializing these inventions. Right now, we are at Version 3, which is more similar to a word processor, led by Prime Medicine, Tome, among others.
The history of modern gene editing, after 2012, has been anchored around patents. And the series of lawsuits & subsequent settlements. But once a company can get clean CRISPR IP, then success really becomes about target-product profiles. Platforms in the field are interesting but CRISPR proteins are a commodity. It’s more about your ability to refine them into products. And like any gold rush, infrastructure matters a lot more. Any panhandler can show up and claim a stake but it takes a serious group to commercialize this technology. Similarly, the CRISPR companies that will leave a lasting legacy will develop successful medicines regardless if they are using version 1, 2, or 3 and so on technologies.
The 2012 Science paper was truly revolutionary. It lowered the barriers to create knock-outs, generate transgenic models, and so much more. I was doing summer research around this time and was so happy to not have to touch TALENS again. Cloning and PCR can be pretty hard. Instead of taking a year to clone a TALENS, CRISPR was a 2 part system that can be ordered off-the-shelf and programmed to target a pretty large set of the genome. The first version had issues with on/off-target effects, insertion/deletions (indels), protospacer adjacent motifs (PAM), delivery, and immunogenicity. While CRISPR proteins have become more specific, the original issues around delivering them to specific organs/tissues and immunogenicity are still outstanding problems to solve.
Before expanding on each version of CRISPR (1 to 3), it’s important to lay out the various characters. Progress in the field has been driven mainly by two camps: one led by Jennifer Doudna and the other led by Feng Zhang, who did foundational work to apply CRISPR to mammalian cells and in my opinion will win a Nobel Prize sooner or later. Likely for his graduate research developing optogenetics. Both groups have attracted and developed some of the most talented individuals in biotech right now. Their progeny have helped get various companies off-the-ground along with setting up their own labs to expand the CRISPR toolkit.
The Doudna lineage starts with Kaihong Zhou (lab manager who has been with Jennifer from the start), in my opinion because she is the lab’s secret weapon. Kaihong keeps the lab running smoothly and helped lay the foundation for them to become the leader in establishing new discoveries for CRISPR & setting the pace for many new approaches. Rachel Haurwitz, Stanley Qi, and Martin Jinek (lead author on the 2012 paper) were part of the first wave of Doudna-lab alumni post-CRISPR. Rachel co-founded Caribou Biosciences. Stanley became a professor at Stanford and recently got Epic Bio off-the-ground. And Martin is a professor at the University of Zurich. The second wave had Ben Oakes (Scribe Biosciences), Janice Chen & Lucas Harrington (Mammoth Biosciences), Mitch O’Connell (Rochester), Alexandra Seletsky (Arrakis Therapeutics), James Nunez (Berkeley professor), Brian Thomas (Metagenomi), Ross Wilson (Berkeley) - truly a special class. The current wave from the lab includes Macro Lobba (Catena Biosciences), Shakked Halperin (Rewrite Therapeutics), and Basem Al-Shayeb.
The Zhang lineage leans toward tool development. Patrick Hsu, now a professor at Berkeley, was early at Editas and is currently involved in a diverse set of gene editing companies. Jonathan Gootenberg and Omar Abudayyeh (i.e. Jomar) are currently McGovern fellows at MIT - it is extremely rare for two great scientists to team up for a joint lab and share credit. The only other comparables that come to mind are Goldstein & Brown and Ruvkun & Ambros. Jomar co-founded Tome and several other companies; they are a golden goose. Winston Yan and David Scott co-founded Arbor Biotechnologies. Many Zhang alumni started their own labs like Neville Sanjana (NYGC), Randall Platt (ETH Zurich), Jonathan Schmid-Burgk (Bonn), and Sidi Chen (Yale). Or joined new companies like Ian Slaymaker (Beam Therapeutics). The new wave from the Zhang group includes Julia Joung (postdoc in the Jonathan Weissman lab at UCSF), James Briggs, Michael Segel, Han Altae-Tran, and Soumya Kannan. Beyond these 2 lineages, other pioneers are David Liu (inventor of base & prime editing), Matthew Porteus, Nicole Gaudelli, Alexis Komor, J. Keith Joung, Luciano Marraffini, Ben Kleinstiver, Jonathan Weissman, George Church, among others. It has been a world-wide effort to expand upon the 2012 discovery and create a golden age in gene editing.
Each version of CRISPR builds upon the last. Work between 2012 and 2016 mainly dealt with effector selection and the corresponding PAM compatibility. CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) is actually a bacterial defense system against bacteriophages and has two major classes:
Class 1: Uses multiple protein complexes to cleave a nucleic acid
Class 2: Uses one protein domain to initiate cleavage. CRISPR effectors used in drug development and research often come from this class due to their simplicity. Within this class there are three main types with their own effector - type II (Cas9), type V (Cas12; I preferred the original Cpf1 name), and type VI (Cas13). Cas9/12 target DNA while Cas13 goes after RNA. In general, Cas12 variants have lower off-target edits than Cas9. There are many more subtypes to choose from and interesting effectors in between like CasX.
Once an effector is chosen, you have to figure out how to get it to work in a specific cell type (i.e. dividing versus postmitotic), form factor (i.e. mRNA, plasmid DNA, virus, ribonucleoprotein), and delivery vehicle (i.e. LNP, AAV, electroporation). These are important decisions; for example, using a ribonucleoprotein (RNP) complex often leads to lower off-target edits. Depending on whether you are correcting a gene or disrupting it along with cutting it or not, the mixture of indels becomes an important metric (ideally less than <0.01%) before moving forward into the clinic. The first version of CRISPR relies on cutting DNA and repairing it through homology directed repair (HDR). This leads to quite a high number of indels, sometimes well over 1%. Overall a lot of protein engineering is done to increase the fidelity of this process and expand the target scope for various CRISPR effectors. More work is needed to discover and engineer higher fidelity, reducing off-target edits while maintaining on-target activity and guide RNA compatibility. Lastly, immunogenicity is an existential risk for all CRISPR programs. Issues have not arisen so far in clinical trials but the field is still early. Interesting work to discover nucleases such as Cas14 and CasΦ in non-pathogenic microbes might be able to sidestep this risk.
Fidelity becomes the most important feature to make CRISPR into medicines. Targeting requires a PAM, which has to be near the target DNA site. The very first effector, SpCas9, relies on an NGG (3 nucleotides where N can be any of the 4) PAM that must be 15±2 bases from the target. For SpCas9, this means that the effector can address about 25% of pathogenic point mutations. Other naturally-occurring CRISPR proteins expand the menu for PAMs and increase the coverage of addressable mutations. Then they can be engineered/evolved to broaden PAM compatibility. In 2015, work from J. Keith Joung’s lab led by Ben Kleinstiver invented 3 SpCas9 mutants each with a unique PAM: SpCas9-EQR recognizing NGAG, VQR for NGA, and the VRER mutant for NGCC. The David Liu lab also has done pioneering work to apply directed evolution to CRISPR and expand their PAM recognition capabilities.
After the 2012 paper, base editing was the next key milestone in the field. Based on research from David Liu’s lab in 2016 & 2017, base editors enabled point mutation correction without requiring a donor template or double-stranded breaks (DSB) in DNA. The 2016 paper invented the first cytosine base editor (CBE) to convert targeted cytosine-guanine (C-G) base pairs into thymine-adenine (T-A) base pairs. Then the 2017 publication built upon this by inventing the first adenine base editor (ABE), which converts A-T to G-C. If the first version of CRISPR was a pair of scissors, base editing would be similar to an eraser. The former would induce DSBs and open up the possibility of pieces of DNA being inserted or deleted (indels) and significantly altering the sequence of the gene. By avoiding cuts, base editors became higher fidelity tools for point mutations with 40%-50% efficiency rates with indel rates below 1%.
Version 3 of gene editing is based on more easily inserting and removing large segments of DNA sequences. Prime editing from David Liu’s lab (published in 2019) established the field and recent work to invent PASTE (relies on integrases) from the Abudayyeh-Gootenberg (AbuGoot) lab and discover new recombinases from Patrick Hsu’s lab builds upon it.
After a version of CRISPR is chosen, it’s important to match the edit to the effector. Each one has their set of advantages and disadvantages based on their function:
Base editor = Cas9 linked to deaminase
Prime editor = Cas9 linked to a reverse transcriptase
CRISPR effectors are linked to integrases for insertion
Overall, gene editing will first address diseases with highly penetrant mutations. Like sickle cell disease (SCD), Huntington’s disease, and many in the liver (due to ease-of-delivery). The wide set of tools developed help expand gene editing’s purview to pursue the over 70K pathogenic genetic variants across about 7,000 known genetic diseases affecting 100Ms of people along with their families:
30% = transition point mutations (C->T, G->A, A->G, T->C)
~25% = deletions
20% = transversion point mutations (8 others)
~8% = duplications
The rest are copy number gain/loss, insertions, and more
Each version of CRISPR will play an important role in treating genetic diseases. The type of edit will determine which effector is used. Canonical effectors will be useful for penetrant mutations. Base editing for transition point mutations. Prime editing for transversions. Integrase-based tools for deletions. Each candidate will be matched to a disease (i.e. target-product profile) by balancing editing efficiencies and insertion/deletion (indel) byproducts with targeting flexibility. For example, base editing, which relies on a deaminase to convert base pairs (4 transitions) has fewer indels and higher efficiency versus prime editing that uses a reverse transcriptase to replace DNA sequences from a template. However, the latter has a much higher targeting flexibility than base editing and can address transversion point mutations. Various analyses are done to match the right effector and editing approach to an indication.
Over the last 10 years, we now have the ability to modify bases, insert/delete genes, edit RNA and the epigenome, edit the mitochondrial genome, develop rapid diagnostics, and more. New CRISPR variants will be discovered and unlock more applications. The last decade has shown that nature has invented countless CRISPR proteins and have made them into a commodity. Matching them to the right edit and disease is where most of the value is created. Less so on the discovery side. Fidelity and effector/edit matching are the key drivers for success. But once this upfront work can be done, the easier programmability of CRISPR should enable efficient ways to have a large swath of genetic diseases potentially cured.
“CRISPR technologies are powerful gene editing tools because they are programmable.”
- Feng Zhang, Professor at MIT and pioneer in gene editing