
Axial Discovery - Immunotherapies and the Undruggable Genome
Axial Discovery - Immunotherapies and the Undruggable Genome
Call for talented individuals and teams

Analysis sent out every Monday
Build with Axial: https://axial22.axialvc.com/
Axial partners with great founders and inventors. We invest in early-stage life sciences companies such as Appia Bio, Seranova Bio, Delix Therapeutics, Simcha Therapeutics, among others often when they are no more than an idea. We are fanatical about helping the rare inventor who is compelled to build their own enduring business. If you or someone you know has a great idea or company in life sciences, Axial would be excited to get to know you and possibly invest in your vision and company . We are excited to be in business with you - email us at info@axialvc.com

Immunotherapies and the Undruggable Genome
Recent work out of UCSF has figured out a way to guide the immune system to target cancer cells that have certain mutated versions of proteins currently considered undruggable. Versus directly drugging these proteins, the paper invents an alternative route to use an immunotherapy to go after them and circumvent the challenges of developing chemical matter that need to directly engage these targets & are also drug-like. This is probably one of the most important research papers published this year.
The 3 key figures for this work are Kevan Shokat, Charles Craik, and Ziyang Zhang. Kevan, a legend in cancer drug development. Charles, a legend in his own right pioneering work in chemical biology and protein engineering. And Ziyang, a rising chemistry star. Kevan brought decades worth of expertise in chemical biology. The work built upon past discoveries around KRAS (mutated in around 20% of all cancers) his lab made that ultimately led to the approval of Lumakras. Charles brought his group’s antibody engineering capabilities. Then Ziyang merged both together at the intersection of chemistry and immunology to invent a new paradigm for cancer drug development.
Kevan Shokat grew up in East Bay around Berkeley and Albany. His initial interest in chemistry was influenced by his parents’ printing business where he practiced a lot of chemistry without even knowing it. At Reed College, Kevan actually took courses leaning toward biology, maybe even pre-med. After an organic chemistry class, he realized a lot of the ink mixing he had done growing up was all chemistry. Motivating Shokat to switch majors and focus on chemisty. On a side note, orgo classes are some of the toughest college courses but at least for me, the most rewarding. Eric Jacobsen taught me one half of organic chemistry, and he would draw out reactions on the chalkboard with both of his hands, and imprinted on me that all of nature has a common design system: giving electrons a path to go one way or another.
After studying chemistry at Reed, Kevan joined Peter Schultz’s lab, one of the best chemists ever and a pioneering translational researcher around the applications of genetic code expansion, at UC Berkeley for his PhD. His graduate research was focused on using antibodies as enzymes and honing the specificities of these catalytic antibodies. A key decision in his career was diversifying into immunology. I remember Craig Crews telling me at a dinner, a key trait of any great scientist is showing success in 2 separate fields. After practicing chemistry under one of the best chemists ever, Kevan was looking for a new opportunity in the Bay Area because his wife started her residency at UCSF. He ended up with a postdoc at Stanford in Chris Goodnow’s lab.
At Stanford, Kevan’s work revolved around B-cells and kinases. After overcoming the initial hurdle of learning immunology & proper design of corresponding experiments, he started developing chemical tools to ID immunological targets. His work at Stanford set him on his current research path of using chemical genetics to study signaling proteins. Setting up his lab at Princeton in 1994 and moving over to UCSF in 1999, Shokat’s group was at the forefront of chemical biology, developing ATP analogs to label kinase substrates and map signaling networks. This work evolved toward developing selective molecules that inhibit one kinase while avoiding ones with similar binding pockets.
The key piece of work in his career was a culmination of decades of research across chemical biology. KRAS, a well-known undruggable target in cancer & one of the Four Horsemen in the field (KRAS, c-MYC, BRAF, p53), caught the Shokat lab’s attention after having the idea to screen for chemical matter against the G12C mutant version of KRAS. Cysteines (C) have a sulfur on its side chain, which are highly reactive with electrophiles to form covalent bonds. An initial screen discovered a new binding pocket in an OFF-state of KRAS leading them to develop a candidate drug that interacts with the site and stops/reduces KRAS(G12C) activity. The key breakthrough for this work was using an acrylamide electrophile in their screening library with reactivity that is location specific given that G12C and not G13C could be covalently modified. Likely due to G12C’s proximity to other side chains. Acrylamides are the “Goldilocks” electrophile and still popular because the rules that determine their reactivity are well known & can be added to a hit later on in the synthesis process. For example, Principia Biopharma also used the cyano version of the functional group in their BTK inhibitor, rilzabrutinib. Ultimately, the Shokat lab’s work was published in 2013 and led to the ultimate approval of Lumakras (sotorasib) in 2021.
Charles Craik joined UCSF in 1985. He is actually trained as a chemist and actually had to give up a basketball scholarship in college to spend more time studying chemistry. His initial work was around structural biology to develop inhibitors of HIV protease. Over time his lab expanded to antibody engineering, substrate profiling, and even imaging. As a collaborative group, the Craik lab’s work touched upon infectious disease, cancer, and neuroscience. Ziyang Zhang, who recently joined Berkeley as a professor, is an emerging inventor at the intersection of chemistry and immunology. Zhang trained under Andrew Myers whose chemistry tree is as legendary as Bill Walsh’s coaching tree, working on total synthesis of macrolides. Zhang then joined the Shokat lab for his postdoc to develop new ways to drug various mutant variants of KRAS. Beyond the G12C mutation, there are also G12D and G12V versions that are much harder to drug with covalent inhibitors due to their less reactive side chains. In 2020, he was the lead author for work that discovered candidates against KRAS(G12D). His key work published this year worked with the Craik lab led by Peter Rohweder to use their antibody engineering capabilities and combine them with a covalent strategy to display fragments of KRAS to the immune system. This led to a new strategy to guide the immune system to attract cancer cells with the KRAS(G12C) mutation.
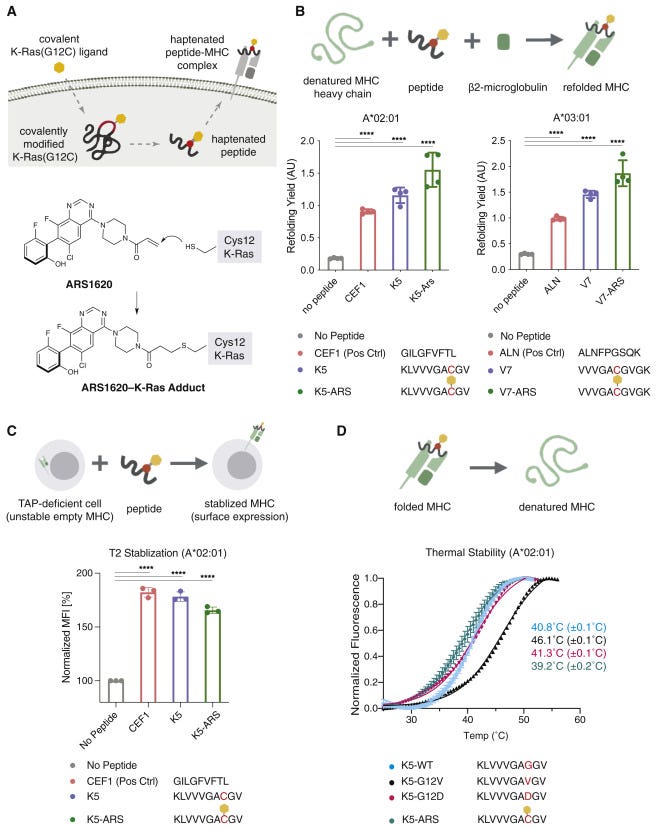
This work is decades in the making. And builds on Shokat’s covalent KRAS(G12C) inhibitors. The key idea was to use a covalent inhibitor as a hapten to generate modified KRAS neoepitopes. The paper then developed antibodies against these epitopes with phase display that induce T-cell mediated killing of cancer cells with the G12C mutation. Fragments of KRAS and other undrugged targets have been observed to be presented by major histocompatibility complex (MHC). However, developing antibodies for antigens with single amino acid substitutions is pretty tricky. So the group explored where covalent modified fragments of KRAS(G12C) would get displayed and recognized by the immune system. So once they found that MHC displayed KRAS fragments tagged by a version of sotorasib, ARS-1620, the race was on to discover an antibody for the protein:small molecule complex.
No longer hiding within the cell, KRAS fragments exposed extracellularly can be targeted by an antibody. That’s where Craik’s team comes in. Using phage display, an initial antibody was discovered. Then a bispecific T-cell engager (BiTE) was designed from the initial antibody clone. However, it also bound to free ARS-1620 before interacting with KRAS - a new antibody was engineered to improve selectively for the KRAS/ARS-1620 complex, but the paper relied on the original antibody clone. They then established that they could induce T-cell killing in cancer cells treated with ARS-1620 + the antibody. The key experiment was showing that the treatment program could kill cancer cells resistant to ARS-1620. Alluding to a potential for an immunotherapy used against cancer resistant to sotorasib and other drugs. However, work is being done to validate this research in vivo, which will unveil new problems to solve before bringing this research closer to the clinic. The long-term plan is to generalize this strategy of moving fragments of undrugged proteins to the cell surface and make them amenable to an immunotherapy. Ultimately, the paper adds a 3rd way to drug cancer: one approach is to develop targeted drugs and another is to make therapies that unleash the immune system. Zhang, Craik, and Shokat invented an ability to use both to expand the scope of immunotherapy to undrugged targets and develop medicines that could potentially retain efficacy even if the cancer becomes resistant to direct drug inhibition.
From the paper, the key experiments were establishing MHC recognition, antibody screening, complex formation/display, and finally T-cell killing:
Two MHC class I alleles, HLA-A∗02:01/03:01 had been observed to contain KRAS peptide epitopes. So 2 peptides were made for both by solid-phase synthesis and conjugated to ARS-1620 with a Michael addition. The modified peptides were found to form complexes with the MHC proteins detected by an ELISA.
The paper used a B-cell derived Fab phage display library with ~10^10 clones. Antibodies that bound to the ARS-1620 modified peptide were selected for and pulled down with a biotin tag. A negative selection was done to filter out antibodies that bind the unmodified peptide. Four rounds of selection generated 186 clones that were then honed down to 5 that showed affinities between 14 to 51 nM.
The P1A4 clone was chosen from the 5 due to its shorter CDR3 loop that might make space for the hapten. The antibody was found to bind T2 cell lines after treatment with the peptide conjugate (K5-ARS), and was immunoprecipitated from 3 KRAS(G12C) cell lines to measure P1A4’s association. A proximity ligation assay was also used to measure ARS-1620/MHC co-localization. Then in mouse xenografts treated with ARS-1620, P1A4 stained for the cancer cells and was treatment selective. Good molecular biology designs experiments to triangulate. These experiments triangulated to confirm that P1A4 recognizes the MHC-displayed peptide.
Lastly, the paper treated a cell line with ARS-1620, 10 μM concentration over 14 days to create a resistant cell to treat with a P1A4xCD3 bispecific (10 nM). After treatment, PBMCs were able to kill the cancer cells or reduce growth inhibition at a ~69% rate. In a clinically relevant cell line with a G12V KRAS variant, the bispecific induced a ~77% inhibition rate.
Beyond KRAS, the next set of targets that are obvious to pursue with this strategy are TP53 (Y220C), GNAS (R201C), and IDH1 (R132C), which all have cysteine residues and are associated with cancer drug resistance. More work needs to be done on developing bispecifics with a higher affinity for the hapten:target complex then generating data in animal models and expanding the approach to more haptens, proteins, and cancer cell types.
High-throughput proteomics has already enabled the development of more selective covalent inhibitors. And this toolkit will be essential to systematize hapten/antibody combination cancer immunotherapies. By the 2010s Shokat’s work along with research from the labs of Ben Cravatt, Daniel Nomura, among others established the toolbox for covalent medicines. A proteomic screening workflow can profile (ABPP) chemical fragments that bind a target(s) and their off-target effects. Before current proteomic methods, this type of work was more like a shot in the dark. So instead of engaging hundreds of targets, a covalent inhibitor that interacts with maybe 10s at most can be discovered. As a result, the current generation of covalent inhibitors will be designed with polypharmacology in mind. For example, the Shokat lab has recently done work around here for PI3Ks and mTOR. And the startup field is led by Meliora Therapeutics to develop drug candidates that engage multiple targets and achieve outcomes not possible with single-target therapeutics.
The other opportunity is to expand the possible residues for covalency beyond cysteine. To serines, lysines, methionines, aspartate and tyrosines. The initial KRAS inhibitors developed by Shokat’s group relied on acrylamide reactive groups with just good enough affinity but high cysteine reactivity. Expansion will require moving beyond acrylamides. Reactive groups like epoxides and boronic acid (for threonine) are starts. This is a space in need of creative chemistry that works in water. On top of this, electrophile-first drug screening campaigns are a major shift in thinking over the last decade. With the clinical validation of covalent inhibitors and the rise of proteomics & activity based protein profiling, ligandable sites can be discovered systematically and offer new starting points for drugs. With around 80% of all proteins still undrugged, new strategies are needed. The paper Shokat, Craik, and Zhang published builds on top of the progress in covalent drugs over the last decade or two and uses it to expand the applicability of immunotherapies. With these two waves meeting at this research paper, multiplicative effects emerge to provide more options for drug developers to treat cancer.
“I think 50–100 years from now, there will be no protein that is undruggable. Everything will be druggable.”